
医疗器械是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件。
医疗器械作为医药行业的重要细分领域,越来越受到国家的重视,近年来政府不断推出有效的政策和措施推动医疗器械产业发展,我国相关政策法规正逐步改进和完善。以下总结了欧盟及美国有关医疗器械法规,与大家进行分享。
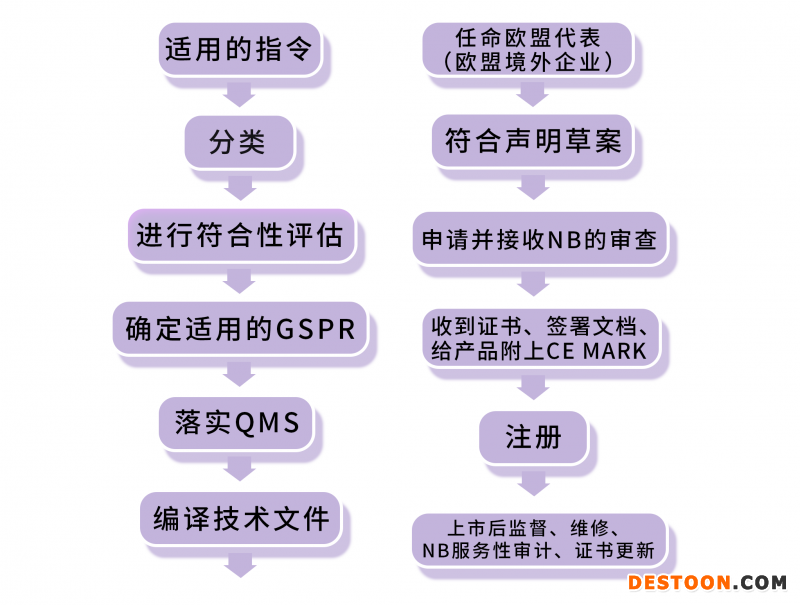
(MDD-MDA)
考虑到器械的预期用途及其固有风险,器械应分为I、IIa、IIb和III类。
所有非创伤性器械均属于I类,除非他们:
-
用于储存体液(血袋例外) IIa类
-
于II a类或更高类型的有源医疗器械类 IIa类
-
改变体液成分 IIa/IIb类
-
一些伤口敷料 IIa/IIb类
-
用于储存体液(血袋例外) IIa类
-
于IIa类或更高类型的有源医疗器械类 IIa类
-
改变体液成分 IIa/IIb类
-
一些伤口敷料 IIa/IIb类
-
再使用的外科器械(钳子,斧子) I类
-
暂时或短期使用(缝合针,外科手套) IIa类
-
长期使用(假关节,眼内晶体) IIb类
-
与中央循环系统(CCS)或中枢神经系统接触的器械 III类
-
给予或交换能量的治疗器械 IIa类
(肌肉刺激器、电钻、皮肤光疗机、助听器)
-
一种潜在危险方式工作的 IIb类
(婴儿培养箱、高频电刀、超声碎石机、X光机)
-
提供能量(核磁共振,超声诊断仪)IIa类
-
诊断/监视体内放射药物分布 (r照相机、正电子发射成像仪) IIa类
-
诊断/监视生理功能(心电图、脑电图)IIa类
-
危险情况下监视生理功能 (手术中的血气分析仪) IIb类
-
发出电离辐射(X射线诊断仪) IIb类
-
控制药物或其他物质进出人体的有源器械 II a类 (吸引设备、供给泵)
-
以一种潜在危险方式工作 IIb类 (麻醉机、呼吸机、透析机、高压氧舱)
-
所有其他有源医疗器械属于I类
(观察灯、牙科椅、轮椅、牙科用治疗灯、记录处理观察诊断图象用的有源器械)
-
与医用物质结合的器械(含杀精子的避孕套、含抗生素的牙髓材料) III类
-
普通Ⅲ类和Ⅱb类产品:NB将审核报告提交给expert panel,21天内给出反馈意 见,如果60天内还没有任何回复,默认是同意的;
-
含药器械:含药部分需要药物主管当局或者EMA(欧洲药品管理局)审核,官方 的时间为210天(Directive 2001/83/EC );
-
动物源性产品:NB应该按照Regulation (EU) No 722/2012的要求进行审核;
-
可吸收或降解产品:关于可吸收或者降解部分需要结合相关当局或者EMA的意见,官方时间为150天(Directive 2001/83/EC)
-
人源的组织或细胞或其衍生物产品:人源的组织或细胞或其衍生物部分需要结合相 关当局的意见,官方时间为120天(Directive 2004/23/EC)。
临床评价应当遵循明确的、良好的方法论并基于:
-
对和器械安全性、性能、设计特性和预期用途相关的现有文献的批判性评价, 并满足以下条件:
(1)临床评价器械的预期用途和数据相关器械的等同得到证实;
(2)数据充分证实了对相关通用安全及性能要求的符合性
-
对所有临床试验结果的批判性评价;
-
考虑现有的其他治疗方案。
临床评价是一个收集和评价与器械相关的所有临床数据的过程,并评价是否有足 够的临床证据证明符合相关的法规要求。临床评价报告中记录了这一过程及其结果。
美国FDA注册申报
要求医疗器械上市前需向FDA提交证明,该装置在市场上销售至少是安全和有效的。提交者必须比较一个或更多的类似合法上市的设备,使他们的设备和支持他们的其他设备实质性等同。
一些I类和大多数II类器械需要510(k)。在510(k)中,根据需要,提案人必须证明新器械在预期用途,技术特性和性能测试方面与判定器械“基本上等同”。
大多数III类器械需要PMA。PMA是最严格的上市前提交类型。在FDA批准PMA之前,申办者必须提供有效的科学证据,证明对器械的预期用途的安全性和有效性的合理保证。
De Novo为新器械提供了一种手段,没有有效判定的情况下,如果符合某些标准,则被分类为I类或II类。
HDE为第III类器械提供了旨在使患有罕见疾病或病症的患者受益的调节路径。为了使器械符合HDE资格,提案人必须获得指定为人道主义使用器械(HUD),该器械是通过向FDA孤儿产品开发办公室(OOPD)申请获得的。
-END-





